Regardless of what type of medical device you develop, the FDA requires you to follow a regulated process. Each step of medical device design lays the foundation for the next stage resulting in a product that meets the user’s needs and is safe and effective. In this guide, we will explore each step in detail so you can understand the complexities of medical device development. Our goal is that by following the steps in this guide, you can design a medical device that meets user needs, complies with FDA regulatory requirements, and is safe and effective.
1. Define the intended use
At Synectic, we recommend first identifying your medical device’s intended use. Your intended use determines your medical device’s regulatory oversight, including testing data, regulatory approval pathway, and classification necessary to gain FDA approval. The FDA defines the intended use of a medical device as the “general purpose for which the device is intended to be used, and the specific uses for which it is labeled or advertised” (21 CFR 801.4). Our regulatory engineers manage V&V planning and execution as part of every medical device engagement.
As stated above, intended use drives your regulatory pathway. Unfortunately, we have seen firsthand the results of a medical device company improperly identifying the intended use. As a result, you can spend lots of time and money going down a specific pathway only to have the FDA reject your 510(k) application because of misidentification. Here is how we recommend you go about identifying your medical device’s intended use:
- Start by defining the medical condition or problem your device intends to address. For example, if you are developing a medical device for diabetes, the problem your device addresses would be “to measure blood glucose levels in patients with diabetes.”
- Once you have defined the medical condition or problem, identify the intended patient population. In our blood glucose measuring device example above, the patient population would be “patients with gestational diabetes.”
- Describe the device’s intended purpose. For example, the intended purpose would be “to measure blood glucose levels as needed using a finger prick test.”
- Define any limitations or contraindications, including specific patient populations or adverse conditions. For example, “not for use in neonates or critically ill patients.”
- Consider any other factors impacting the intended use, such as the environment. For example, “not to be used in over 80% humidity.”
2. Classify your device
After identifying your device’s intended use, you can classify your device by reviewing the FDA’s classification database. As mentioned earlier, your device’s classification determines the amount of regulatory oversight required for approval. It is also important to note that the intended use and patient and user risk affect classification. For example, if a general-use device will now save someone’s life, the level of risk changes and, therefore, the classification level. The FDA classifies medical devices into three categories based on their level of risk:
Class I
Class II
Class II medical devices are moderate-risk devices like infusion pumps and diagnostic imaging equipment. Class II devices are subject to premarket notification (510(k)), which requires the manufacturer to demonstrate that the device is substantially equivalent to a legally marketed device.
Class III
Medical devices classified as Class III include implantable devices and those that support or sustain life. Class III devices pose the greatest risk and are subject to the most regulatory controls. They require premarket approval (PMA), which includes a comprehensive clinical and scientific data review to demonstrate safety and effectiveness.
3. Develop a Quality Management System (QMS)
As a medical device manufacturer, you must create a quality management system (QMS). A QMS is a set of risk management procedures and controls that guarantee your medical device is safe and effective. The FDA outlines the requirements for a QMS in the Quality System Regulation (QSR), also known as 21 CFR Part 820. The QSR details what current good manufacturing practices (cGMPs) you should follow, including:
- A management structure responsible for establishing, implementing, and maintaining the QMS
- Procedures for implementing medical device design controls so that your medical devices meet all applicable regulatory requirements
- Procedures for maintaining and controlling quality-related documents such as SOPs, work instructions, and design verifications
- Procedures for controlling the approval of vendors and purchasing materials
- Procedures for controlling production and manufacturing processes
- Procedures for identifying, investigating, and correcting nonconformities arising from the manufacturing process
- Procedures for ensuring your medical devices are correctly packaged and labeled
- Procedures for handling, storing, distributing, and installing your medical devices properly so no damage or degradation affects safety and reliability
- Procedures for receiving, reviewing, and evaluating product complaints
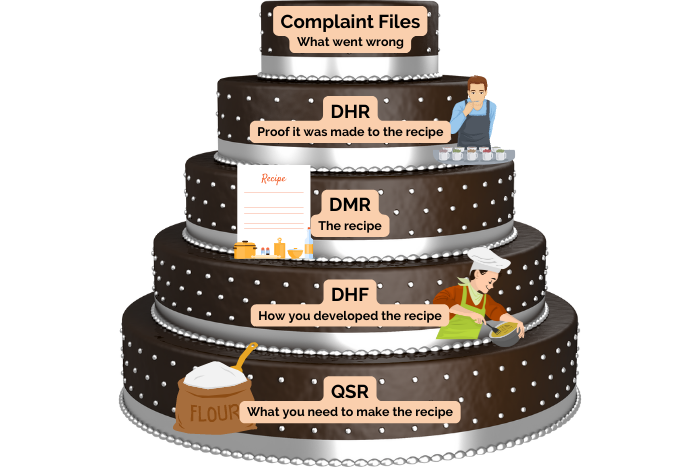
- Procedures for maintaining records related to the QMS and the manufacturing process, such as the design history file (DHF), device master record (DMR), device history record (DHR), quality system record (QSR), and complaint files
4. List the user requirements
User requirements describe the characteristics and features that your device must have to address the intended use. To determine your user requirements, engage with potential patients, clinicians, and other users to gather feedback and insights. You should also review the target user group, the device’s intended use, and the regulatory requirements.
At Synectic, we compile user requirements in the product development specification (PDS) document. The PDS is a written document that directly aligns the user requirement and design input to the design output and verification activity. As a result, the PDS makes it possible to quickly assess which design inputs you have satisfactorily met, and which design inputs still need to be evaluated or verified. Repeated use of the PDS can also provide a history showing the design meets the requirements.
Remember that user requirements are not solutions. They should remain broad, so you do not hinder the design process. If you start with specific technical requirements, you might pigeonhole yourself into a design trajectory that fails to address the intended use. An example of a user requirement could be “handle must be blue” or “usable one-handed.”
5. Identify the design inputs
The design inputs are where you translate your general user requirements into technical specifications and criteria that guide your medical product design. Design inputs provide a clear and detailed description of the device’s features, functions, and performance requirements, serving as a roadmap for the design team. Throughout medical device product development, you will create design outputs that meet each design input and use verification activities to confirm this.
When identifying your design inputs, match each user requirement with at least one technical design requirement. For instance, if the user requirement is a blue handle, the corresponding design input would be the specific Pantone color, which can be verified using a mass spectrometer. Design inputs may include the following criteria:
- Size and weight
- Cost
- Usability
- Biocompatibility and sterility
- Durability
- Performance
- Safety and regulatory
- Environmental and operational conditions
- Packaging and Labeling
6. Design your medical device
Engineering medical devices involve creating detailed drawings and models of the device, including all necessary components and materials. However, before you dive into sketching, CAD work, and prototyping required for medical device design, there are several things you must first consider.
Human-factors engineering
Human factors engineering is the application of psychological and physiological principles to medical devices to optimize their interaction with people. It is critical to ensure the safety and effectiveness of medical devices, as well as clinician and end-user satisfaction and comfort. Human factors engineering is so important for ensuring medical product designs minimize the risk of errors or injuries that the FDA has established guidelines for it.
During medical device development, you should consider how the device will be used in real-world conditions and design for usability and ease of maintenance. Human factors such as user error, environmental conditions, and device maintenance can contribute to failure modes and design weaknesses.
Accessible and inclusive design
When designing medical devices, remember they must be accessible and inclusive to all users, including those with disabilities. You can do this by incorporating several design features, such as:
- Using large, easy-to-read fonts and high-contrast colors for easy readability
- Avoiding using color as the sole means of conveying information so your medical device is usable by those with color blindness
- Including visual cues for the deaf and hard of hearing, such as flashing lights or vibrating alerts
- Incorporating large buttons or touchscreens to make it easier for motor-impaired users
Designing for safety and reliability
Remember that the entire reason the FDA regulates medical device development is to provide the market with safe and reliable devices. Poorly designed medical devices can lead to patient harm and even death. When developing medical devices, we recommend conducting finite element analysis, simulations, and other tests to lower risk. By performing risk management activities, such as a DFMEA, you can identify and address potential failure modes, design weaknesses, and risks associated with the device. We suggest the following risk management activities during medical device product development:
- Perform a risk analysis to identify potential hazards and develop mitigation strategies.
- Include redundant systems in your design to ensure the device will continue functioning even if a component fails.
- Incorporate ways for the device to fail safely, minimizing the risk of patient harm.
- Perform safety testing, such as animal testing, clinical trials, biocompatibility and sterility testing, and any other testing required by the FDA.
- Test the manufacturing process and device’s performance and safety under various conditions.
Materials and manufacturing considerations
We recommend you consider materials and manufacturing processes early in the design process. By understanding the limitations and opportunities of various materials and manufacturing processes, you can make more informed decisions about your design concepts’ feasibility and practicality. At Synectic, we consider biocompatibility, cost, durability, sterility, and scalability factors when selecting materials and manufacturing processes.
When selecting materials, make sure you are evaluating them against the device’s requirements. For example, suppose your medical device concept requires a certain level of flexibility while maintaining biocompatibility; you must consider only materials that fulfill both requirements. You may also need to consider materials based on their ability to withstand repeated use or sterilization.
Similarly, the final manufacturing processes can affect the design as each operation must be carefully controlled and validated. Therefore, we recommend selecting manufacturing processes with high quality and consistency—for example, using injection molding or 3D printing for complex geometry parts.
7. Compile your design outputs
Design outputs are the technical specifications, drawings, and documentation that result from the medical device design process. You will use these design outputs to manufacture, test, and validate your medical device. The design outputs are also components of the DHF, which demonstrates regulatory compliance and ensures that the device is traceable throughout its lifecycle. Therefore, any changes to the design outputs must be documented and approved before implementation to confirm that the device continuously meets the user’s needs and regulatory requirements.
Design outputs come in many forms, which can change as the project progresses, such as during design and development; design verifications are often used as design outputs for later phases of the project. For example, if you perform a Finite Element Analysis (FEA) on a plastic hook latch used to secure an access panel, the results of the FEA are considered a design verification. Later, if you create a prototype and test the latch strength, the results of the FEA could be used as a design output to compare against the prototype testing results. Some other examples of design outputs include:
- Technical specifications
- Drawings
- Bill of materials
- Breadboards
- Prototypes
- Manufacturing documents
- Testing procedures and reports
- Engineering calculations
- Tolerance stack-ups
- Work instructions
- Risk management documentation
8. Conduct a design review
A design review evaluates the design’s current state and whether it meets the product requirements. You can perform a design review at any stage of the design and development process. As a result, we recommend performing several throughout the design process to act as benchmarks between major developmental stages. For example, a design review of the CAD work before building prototypes.
Design review activities can change depending on the project’s progress and can include the following:
- Comparing the design progress against the project plan
- Evaluating the design requirements
- Reviewing design outputs
- Verifying that the design outputs meet the design inputs
- Identifying potential problems or risks
- Brainstorming possible solutions or next steps.
Design reviews require attendance by representatives of all functions concerned with the design, such as regulatory, quality, marketing, engineering, and manufacturing. In addition, the FDA also requires attendance by an independent reviewer not directly associated with the design.
During design reviews, participants shall objectively evaluate the design and determine if it is adequate to proceed with the project or if there are design problems to address. A record of the design review, including the date, participants, design identification, results, and necessary actions, shall be maintained in the DHF.
9. Perform verification and validation
Verification and validation are two processes that ensure your medical device meets the user requirements and design inputs outlined at the beginning of the design process. However, while they are related, they are different activities with different goals.
Verification checks whether the device design meets the product specifications. During verification, you compare the design documentation to the product design specification. In short, you are verifying the design outputs match the design inputs and that your processes produce the correct product. Types of verification testing include mechanical, bench, and tissue testing.
Validation checks if the device’s performance meets the intended use and user requirements. It involves testing the device using simulated or actual use scenarios to see if it performs as expected. In short, you are validating that your end product works as intended. Types of validation testing include biocompatibility, clinical trials, and sterility testing.
The FDA outlines specific design control guidelines for verification and validation in 21 CFR 820. Additionally, all verification and validation efforts must be precisely documented and held within the DHF. As a result, we strongly recommend you engage with a regulatory expert to ensure your testing meets regulatory requirements.
10. Obtain FDA approval
To obtain FDA approval, you must prepare and submit a regulatory application with detailed information about your device, its intended use, and its safety and effectiveness data. The type of premarket submission you are required to complete depends on your device classification.
Premarket notification
For Class II devices, you must submit a premarket notification (510(k)) to the FDA. The 510(k) submission demonstrates that your device is substantially equivalent to a legally marketed device (also known as a predicate device) already on the market. The FDA reviews the submission and issues a clearance letter if they deem your medical device substantially equivalent.
Premarket approval
You must submit a premarket approval (PMA) application for Class III devices to the FDA. The PMA application includes comprehensive data demonstrating your medical device is safe and effective such as clinical data from human studies. As with a 510(k) application, the FDA will review your PMA application and issue an approval letter if they deem your medical device safe and effective.
Regardless of which route you take, you will need to demonstrate your device is:
- Safe and that benefits outweigh the risks.
- Effective, and you can support any claims of effectiveness with scientific evidence.
- Manufactured according to current good manufacturing practices (cGMPs).
The regulatory approval process is complex, time-consuming, and requires significant financial resources. While the FDA provides guidance documents and other resources to help you maintain compliance, we recommend working with a medical device design company, like Synectic, that understands how to navigate the regulatory process.
Once the FDA reviews your application, they will clear your device or require you to complete additional steps for approval. If your medical device is approved, you can begin manufacturing and marketing it in the United States.
11. Manufacture your medical device
Medical device manufacturing involves setting up a production process that meets all quality and regulatory requirements and is scalable. As stated earlier, 21 CFR 820.70 states that medical device manufacturers must establish and maintain procedures to control their product design, production, and distribution. Specifically, it requires manufacturers to:
- Ensure the devices are manufactured according to the Device Master Record (DMR) and meet all applicable specifications.
- Maintain calibrated equipment and that all equipment used in production is functioning correctly.
- Test finished devices against the product development specification.
- Establish and maintain procedures for corrective and preventive actions (CAPA), addressing any issues arising during the production process and preventing similar problems.
- Have a way of handling non-conforming products to prevent their release into the market.
12. Conduct post-market surveillance
Compared with regular products, a medical device manufacturer’s job is not done once their device is sold and distributed. The FDA continues to monitor your medical devices’ safety and effectiveness, even after approving them for sale. The FDA regulations around conducting post-market surveillance are found within 21 CFR 822 and include:
Adverse event reporting
As a medical device manufacturer or importer, you must report any incidents where your medical device may have caused or contributed to a death or severe injury. This includes times when the device malfunctioned or did not perform as intended.
Post-market studies
The FDA requires you to conduct post-market studies to collect additional data on your medical devices’ safety and effectiveness. These studies evaluate long-term safety and efficacy and involve observing performance in a real-world setting or conducting clinical trials.
Device recalls
The FDA can order you to recall medical devices that pose a significant patient risk or may ask you to recall voluntarily devices that may pose a patient risk.
Medical device reporting database
The FDA maintains a publicly accessible database of adverse event reports for medical devices called the Manufacturer and User Facility Device Experience (MAUDE) database. This database allows healthcare providers, patients, and manufacturers to report and view incidents where a medical device malfunctioned or caused injury or death.
Inspections
The FDA may inspect medical device manufacturers for compliance with regulatory requirements. These inspections may be routine or in response to specific issues or concerns.
As seen throughout this guide, when designing your medical device, it is better to overanalyze risks than to under analyze and potentially put patients and clinicians in harm’s way. We also strongly suggest you work with a professional medical product design company like Synectic that can guide you through the process. While this guide is long, it is a brief overview of all that medical device design entails. If you want to design a medical device or need help figuring out where to start, contact us below; we would be happy to help.
Need a Development Partner Who Can Own the Full Design Controls Process?
About Synectic Product Development: Synectic Product Development is an ISO 13485 certified, full-scale product development company. Vertically integrated within the Mack Group, our capabilities allow us to take your design from concept all the way to production. With over 40 years of experience in design, development, and manufacturing, we strive for ingenuity, cost-effectiveness, and aesthetics in our designs. Learn more about our medical device design services and see how we can help your next project.