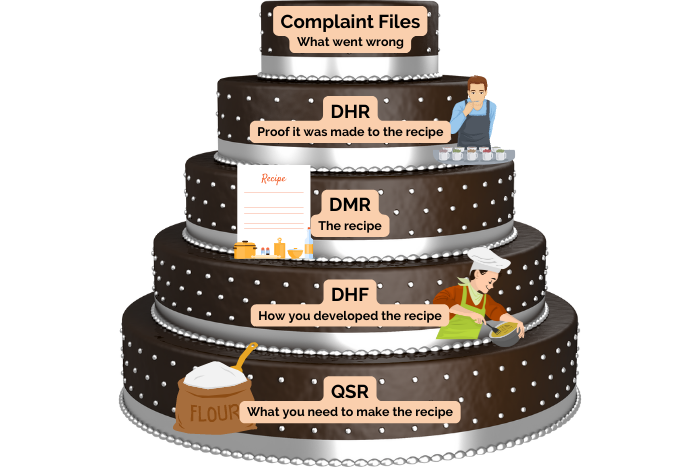
Listed within the FDA’s 21 CFR 820 are detailed regulations governing medical device records and quality systems. These regulations identify five record types that medical device manufacturers need to maintain. These include the Design History File (DHF), Device Master Record (DMR), Device History Record (DHR), Quality System Record (QSR), and Complaint Files. Medical device manufacturers must keep these records for the anticipated lifetime of the device or a minimum of two years from the date of first distribution. Additionally, they must store these records in a way that is easily accessible to anyone within the organization involved in quality-related activities. While the record names may sound similar, they each have a distinct purpose.
Design History File (DHF)
The Design History File is a compilation of records that describe the development history of a finished medical device. It essentially summarizes all actions and thought processes behind device development. When the FDA looks at your Design History File, they want to see that the medical device development happened according to the Design Plan and Quality System Requirements outlined in 21 CFR Part 820. Every type of medical device manufactured requires a separate DHF. Additionally, 510(k) or Premarket Approval requires maintaining a Design History File. The DHF contains:
- Design inputs and outputs
- Design verification and validation
- Design review
- Design plan
- Design transfer
- Risk management documents
Device Master Record (DMR)
The Device Master Record contains all the files necessary to make a completed device. In the simplest of terms, it is a recipe for how to produce your medical device from start to finish. It includes all processes, drawings, labeling requirements, packaging details, and software code. While a lot of the information found in the DMR is also in the DHF, the difference is that the Device Master Record distinguishes the production process from the design process. For example, the DMR includes the instructions for manufacturing a specific part, while the DHF contains part drawings and design outputs. Documents found in the Device Master Record include:
- Device specifications
- Production and process specifications
- Quality assurance procedures
- Packaging and labeling specifications
- Servicing, installation, and maintenance procedures
Device History Record (DHR)
The Device History Record contains files detailing the production history of the finished device. The DHR proves to the FDA that finished devices are manufactured according to the DMR. It lists lot numbers, UPCs, manufacturing dates, quantities, and labeling. A well-maintained Device History Record can trace back each medical device to when, where, and how it was manufactured. If your organization uses a contract manufacturer to produce your finished device, your DHR needs to specify who reviews and releases the finished product. The Device History Record includes:
- Manufacturing date and quantity
- Quantity released for distribution
- Acceptance records demonstrating the device was manufactured correctly according to the DMR
- Labeling for each unit, including signature and date of who examined and released the labels
- Unique Device Identifier (UDI) or Universal Product Code (UPC) and any other identification or control numbers used
Quality System Record (QSR)
The Quality System Record contains or refers to the location of all procedures and records outlined in 21 CFR 820 that are not specific to a particular device. It is essentially everything your organization needs to make the medical device that you could also use to make other devices. The QSR should contain or refer to the following documents:
- Training procedures
- Qualification records
- Internal audit procedures and records
- Management review procedures and records
- Purchasing and supplier assessment procedures and records
- Quality policy and plan
Complaint Files
Complaint files are any communication that alleges deficiencies related to a medical device after releasing it for distribution. The FDA requires manufacturers to have procedures for promptly receiving, reviewing, and evaluating these complaints. Upon reviewing a complaint, the manufacturer must decide whether an investigation is necessary. Complaints involving a novel device or labeling/packaging failure require investigation. Recurring complaints do not require investigation but may need Corrective and Preventative Action (CAPA). If the location of your organization’s complaint unit is outside the United States, the complaint records must be reasonably accessible from within the United States. The regulation regarding complaint files is flexible but at a minimum, they include:
- Device name
- Date of complaint
- UDI, UPC, or any other identification number such as lot or batch numbers
- The complainant’s name, address, and phone number
- Details of the complaint
- Whether an investigation was necessary and the name of the person who made that decision
- Investigation date, if any, and results
- What corrective actions, if any, were taken
- The response to the complainant
To summarize, let us put these medical device record requirements into practice in an everyday situation. Let us say you are developing a new cake recipe. The Design History File (DHF) details how you created the recipe. The Device Master Record (DMR) contains the cake recipe itself. The Device History Record (DHR) is proof that the cake came out according to the recipe. The Quality System Record (QSR) contains everything else you need to make the recipe, such as your oven instruction manual or how to determine if a new flour supplier meets your quality standards. The complaint files are records of what went wrong with the recipe and how you fixed it. For example, if a customer complained that the cake was too dry, you would investigate how this could have occurred and put in a Corrective and Preventative Action (CAPA) to stop it from happening again.
Get a Free Quote
About Synectic Product Development: Synectic Product Development is an ISO 13485 certified, full-scale product development company. Vertically integrated within the Mack Group, our capabilities allow us to take your design from concept to production. With over 40 years of experience in medical device design and manufacturing, we strive for ingenuity, cost-effectiveness, and aesthetics in our designs. Learn more about our medical device design services and see how we can help your next project.